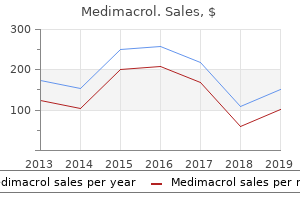
Medimacrol"Discount medimacrol 500 mg on-line, infection 8 weeks after birth". By: H. Mufassa, M.B. B.CH. B.A.O., M.B.B.Ch., Ph.D. Deputy Director, Saint Louis University School of Medicine Once the superficial lobe is mobile the mylohyoid muscle is approached and retracted anteriorly virus united states department of justice buy medimacrol 250 mg with visa. Lying deep to the gland are the lingual nerve superiorly and hypoglossal nerve inferiorly, on the hyoglossus muscle. A small secretorimotor, parasympathetic nerve branch leaves the lingual nerve to enter the submandibular ganglion and then gland. A vessel always accompanies this nerve branch, and must be cauterised and divided to allow mobilisation of the gland with preservation of the lingual nerve. Surgical complications Intraoperative complications Accidental division of the facial nerve should be repaired at the time with a tension-free end-toend anastomosis using the operating microscope. If the tumour is spilt, all macroscopic tumour should be removed and the wound thoroughly irrigated with saline. Facial nerve palsy the risk to the facial nerve may vary with the extent of the surgery, experience of the surgeon, pathology and with recurrent disease. However, reports suggest that they do not reduce the rate of facial nerve palsies. Palatal tumours usually require bony resection and may extend to involve a partial or total maxillectomy. Reconstruction of the defect is usually best delayed until after margins status and adjuvant therapy have been achieved. It is thought to be due to parasympathetic nerve fibres from the auriculotemporal nerve re-anastomosing with sweat glands following parotidectomy. Postoperatively injecting the area with botulinum A is a method frequently employed to reduce symptoms. Good correlation between original and modified House Brackmann facial grading systems. Is ultrasound alone sufficient for imaging superficial lobe benign parotid tumours before surgery A comparative study of the effective radiation doses from cone beam computed tomography and plain radiography for sialography. Utility of immediate on-site cytopathological procurement and evaluation in fine needle aspiration biopsy of head and neck masses. Sonographically guided fine-needle aspiration biopsy of major salivary gland masses: a review of 245 cases. Mumps outbreaks in four universities in the North West of England: prevention, detection and response. Atypical mycobacterial infection of the head and neck in children: a 5-year retrospective review. Nontuberculous lymphadenopathy in children: using the evidence to plan optimal management. Long-term evaluation of extracorporeal shock wave lithotripsy in the treatment of salivary stones. Pleomorphic adenoma of the parotid gland: histopathologic analysis of the capsular characteristics of 218 tumors. Clinical presentation, management, and outcome of high-grade mucoepidermoid carcinoma of the parotid gland. Pathological features of lymphoid proliferations of the salivary glands: lymphoepithelial sialadenitis versus low-grade B-cell lymphoma of the malt type. Primary and metastatic cancer of the parotid: comparison of clinical behavior in 232 cases. Metastases to the major salivary glands from non-head and neck primary malignancies. Surgery of the parotid gland: evolution of techniques, nomenclature and a revised classification system. The precision of four commonly used surgical landmarks for locating the facial nerve in anterograde parotidectomy in humans. Parotidectomy for benign parotid disease at a university teaching hospital: outcome of 963 operations. Nerve paralysis after surgery in the submandibular triangle: review of University of Tokyo Hospital experience. Blood group antibodies are usually IgM or IgG, although some may be IgA (Chapter 6).
The neuronal loss is prominent infection mrsa pictures and symptoms order medimacrol with a visa, although the enzyme is principally expressed in astrocytes and other non-neuronal cells, suggesting impairment of the supply of nutrients derived from metabolic activity in astroglia that are essential for neuronal survival. While subcortical leukodystrophy has developed in some patients, many develop normally and have cognitive ability and motor-skill development that is within the healthy reference range. Diagnosis the condition is suspected when acidosis and neurological disease occur in infants, especially in the presence of hypoglycaemia. Specific diagnosis requires enzymatic assay in fibroblasts, which can also be used for carrier detection. The residual activities appear to correspond to the clinical phenotype approximately. Treatment Episodes of acidosis are treated with intravenous sodium bicarbonate, and glucose may be required for hypoglycaemia. There is evidence that ketogenic diets containing 50% fat and 20% carbohydrate ameliorate the biochemical disturbance and delay the onset of neurological disease. The administration of high-dose glutamate and aspartate, which may act as a source of oxaloacetate, appear to have been beneficial in some patients, at least on the composition of the plasma amino acids. Use of the anaplerotic seven-carbon triheptanoin has been described, with reports of benefit in some cases, but not all. Although biotin therapy has been disappointing in pyruvate carboxylase deficiency, occasional responses to high-dose lipoic acid and thiamine treatment, which may stimulate pyruvate metabolism by the dehydrogenase complex, have been recorded. However, a collaborative effort to investigate their effects in selected patient groups under stratified conditions is needed. Experimental hepatic allotransplantation has been carried out in patients with pyruvate carboxylase deficiency, with salutary effects on plasma amino acids apart from glutamine, but the effect on brain function was not easy to determine in the first patient treated. In: Cornblath M, Schwartz R (eds) Disorders of carbohydrate metabolism in infancy, 3rd edition, pp. Rigor of non-dairy galactose restriction in early childhood, measured by retrospective survey, does not associate with severity of five long-term outcomes quantified in 231 children and adults with classic galactosemia. Genetic basis of transferase-deficient galactosaemia in Ireland and the population history of Irish Travellers. Galactosaemia and allelic variation at the galactose-1phosphate uridyltransferase gene. A re-evaluation of life-long severe galactose restriction for the nutrition management of classic galactosemia. International clinical guideline for the management of classical galactosemia: diagnosis, treatment, and follow-up. Dihydrolipoamide dehydrogenase deficiency: a still overlooked cause of recurrent acute liver failure and Reye-like syndrome. Riboflavin responsive mitochondrial myopathy is a new phenotype of dihydrolipoamide dehydrogenase deficiency. X-linked pyruvate dehydrogenase E1-alpha subunit deficiency in heterozygous females: variable manifestation of the same. A prospective study of growth and nutritional status in children treated with the ketogenic diet. Pyruvate dehydrogenase E2 deficiency: a potentially treatable cause of episodic dystonia. Newborn screening for dihydrolipoamide dehydrogenase deficiency: citrulline as a useful analyte. Evaluation of long-term treatment of children with congenital lactic acidosis with dichloroacetate. All are heterocyclic bases which exist in tri-, di-, and monophosphorylated forms, and as either deoxyribosylated or ribosylated derivatives (deoxyribose and ribose are pentose carbohydrates). Purine compounds also function as signal transducers, neurotransmitters, vasodilators, and mediators of platelet aggregation. Phosphoribosyl pyrophosphate synthetase superactivity presents with uric acid lithiasis or gouty arthritis in childhood or early adult life. Acute attacks of gout are treated with nonsteroidal antiinflammatory drugs, colchicine, or steroids. First-line treatment to prevent acute attacks or manage chronic tophaceous gout is with the xanthine oxidase inhibitor, allopurinol. Other diseases of purine metabolism cause diverse abnormalities and are generally the result of single gene defects, for example, adenosine deaminase and purine nucleoside phosphorylase catalyse sequential steps in the metabolism of purine ribonucleosides and deoxyribonucleosides and are highly expressed in lymphoid cells; their deficiency causes lymphotoxic metabolites to accumulate and leads to lymphopenia and severe combined immunodeficiency. Purchase 100mg medimacrol with visa. HSN | Bzees Footwear 09.21.2017 - 12 AM.
If treatment is begun early in the course of the disease antibiotic z pak purchase medimacrol 250 mg free shipping, neurological and hepatic function can be normalized. However, patients with psychiatric disease may improve, but may be left with residual psychiatric symptoms. Furthermore, advanced disease may not be reversible, and some patients show neurological worsening with initial treatment. If the drug must be withdrawn in the face of side effects or severe complications, attempts can be made to reinstitute therapy, beginning with very low doses, until the therapeutic range is again achieved. Trientine is an effective chelating agent, but experience is limited, and the full spectrum of toxicity is not known. A second alternative drug, ammonium tetrathiomolybdate, has been used primarily as a rapidly acting initial treatment for patients with acute neurological symptoms. These models should be useful for preclinical explorations of novel therapeutic approaches. The effect is mediated by induction of intestinal cell metallothionein, which binds copper with high affinity, preventing the transfer to copper into the blood. Treatment with zinc is nontoxic, does not produce initial neurological worsening, and prevents hepatic accumulation of copper. Treatment with oral zinc has been shown to be effective, with clinical amelioration reported in multiple studies. Oral zinc may be especially useful for maintenance following D-penicillamine initiation, or in conjunction with another agent. In pregnant women being treated with D-penicillamine, the teratogenic effects of the drug must be considered. There are reports of connective tissue defects of variable severity following pregnancies marked by maternal penicillamine use. The magnitude of the risk may be low, as numerous normal babies have been born to mothers treated with D-penicillamine. Maintenance oral zinc therapy may prove to be an important nonteratogenic adjunct during pregnancy. Hsi G and Cox D (2004) A comparison of the mutation spectra of Menkes disease and Wilson disease. Solomon L, Abrams G, Dinner M, and Berhman L (1977) Neonatal abnormalities associated with D-penicillamine treatment during pregnancy. Hepatocyte transfer, an alternative to gene therapy, may also be applicable to the treatment of liver-specific metabolic disorders through therapeutic liver repopulation. Initial partial descriptions of the disorder date from the 1880s, but subsequent developments in clinical and pathological description, metabolism, pathophysiology, genetics, and treatment all occurred during the twentieth century. His definitive publication in Brain in 1912 occupied the entire issue, but his findings were also published in shorter accounts in several languages. Although Wilson was the first to describe the condition in detail, several earlier authors had described such patients. The gene product is expressed heavily in the liver and has been localized to the Golgi complex and possibly to mitochondria. Loss of function of this gene product results in excessive intracellular deposition of copper in hepatocytes, hepatic cellular necrosis, and leakage of copper into the plasma, from whence it is transported to and deposited in other tissues, including the brain. Walshe suggested that penicillamine could be an effective orally administered chelator for copper, a supposition that was soon proven. In a medical thesis in 1961, Dutch neurologist Schouwink demonstrated that orally administered zinc could significantly reduce the absorption of copper from the gut. His inventions included a refractometer for determining refractive indices, a reflecting goniometer to measure angles on crystals, a polarizing beam splitter, and the camera lucida as an optical aid for artists and microscopists. He was elected a fellow of the Royal Society in 1793, and later served as secretary and interim president. In this instance the loss of sight was toward my left, and was the same whether I looked with the right eye or the left. This blindness was not so complete as to amount to absolute blackness, but was a shaded darkness without definite outline. The complaint was of short duration, and in about a quarter of an hour might be said to be wholly gone, having receded with a gradual motion from the centre of vision obliquely upwards toward the left.
Fibrates are the drugs of first choice in the management of severe hypertriglyceridaemia (Table 12 antibiotics for uti didn't work buy medimacrol 100mg lowest price. Others genes, which have yet to be characterized and secondary factors contribute to this phenotype. No single gene has been identified, rather the condition appears to be caused by a combination of gene variants leading to polygenic hypertriglyceridaemia. Acquired (secondary) causes of impaired lipolysis of triglyceride-rich lipoproteins the secondary causes of dyslipidaemia are listed in Table 12. Inheritance of the number of variants together with elevated cholesterol coupled with diet is generally the cause of this condition. In the evaluation of hypercholesterolaemia, however, single-gene Mendelian disorders are relatively common and should be considered in the differential diagnosis (Table 12. These include Afrikaners in South Africa, French Canadians, Christian Lebanese, and the Finns. Secondary causes of marked hypercholesterolaemia, including hypothyroidism, nephrotic syndrome, and obstructive hepatic disease, must be excluded in the differential diagnosis. Hypercholesterolaemia can be detected in the neonate if family history indicates the need for cholesterol measurement. Males have an approximately 50% likelihood of myocardial infarction before the age of 60 and females have an approximately 30% chance. The manifestations of atherosclerotic cardiovascular disease vary greatly in their age of appearance. Bile acid sequestrants can be used before puberty, with other lipid-lowering drugs being introduced after puberty. The rationale for this delay is the importance of cholesterol for growth and sex hormone biosynthesis, but this is not evidence based. The powerful, long-acting statins are the most efficacious drugs and should be used at high intensity in adults (Tables 12. In those planning pregnancy, statins should be stopped, as animal data suggests that statins are teratogenic although there is no human data to support this. When stopping statins prior to conception, a 3-month washout period is often recommended, but this is not evidence based, and shorter periods are probably acceptable. Specific statins and doses are noted in bold that were evaluated in randomized clinical trials, which showed a reduction in major cardiovascular events. Individual responses to statin therapy vary and there might be a biological basis for a poor response. Homozygotes develop atherosclerotic cardiovascular disease in childhood or adolescence. Lesions are common in the aortic root and aortic valve with subvalvular stenosis, often reaching into the coronary ostia, which become narrowed. Without treatment, death is in the second to third decade, reflecting disease severity. Receptor-negative homozygotes are most commonly due to consanguinity, but are fortunately rare. High-intensity statin treatment, ezetimibe, and sequestrants are reasonably given together. Antisense oligonucleotide treatment to block apoB synthesis is used in the United States of America, but not in Europe. It is seen in patients of central European origin and in derived populations in the United States of America, where the frequency is 1 in 500 to 1 in 1000, and this reflects a founder effect mutation. Recurrent mutation occurs as Arg3500 is encoded by a CpG dinucleotide, which is a hotspot for mutation. Cholesteryl ester storage disease Cholesteryl ester storage disease is a rare Mendelian recessive disorder. Patients with no enzyme activity are likely to have the childhood severe presentation and those with partial deficiency the adult presentation. This leads to the accumulation of large amounts of neutral lipid in liver cells and hepatosplenomegaly, with steatosis, fibrosis, and ultimately cirrhosis. It is important to establish the diagnosis and evaluate the liver as cirrhosis may occur. The recommendation was based on four studies which provided evidence on the safety and efficacy in infants (<6 months of age), children, and adults. It is a Mendelian recessive disorder, which becomes manifest when an acquired cause of dyslipidaemia also occurs.
|