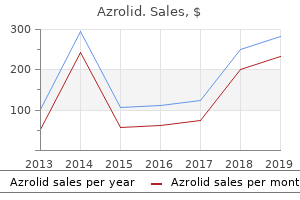
Azrolid"Azrolid 100mg line, antibiotic names". By: W. Dennis, M.A., M.D., M.P.H. Co-Director, Texas Tech University Health Sciences Center School of Medicine Is there altered activity of the extensor muscles in chronic mechanical neck pain? Patients with neck pain demonstrate reduced electromyographic activity of the deep cervical flexor muscles during performance of the craniocervical flexion test antibiotics for uti uk best azrolid 100mg. Patients with chronic neck pain demonstrate altered patterns of muscle activation during performance of a functional upper limb task. Fiber composition and fiber transformations in neck muscles of patients with dysfunction of the cervical spine. Predictive value of fear avoidance in developing chronic neck pain disability: consequences for clinical decision making. A proposed new classification system for whiplash associated disorders-implications for assessment and management. Impairment in the cervical flexors: a comparison of whiplash and insidious onset neck pain patients. Standard scales for measurement of functional outcome for cervical pain or dysfunction: a systematic review. Use of generic versus region-specific functional status measures on patients with cervical spine disorders. Conservative treatment for neck pain: medications, physical therapy, and exercise. Cyclobenzaprine hydrochloride effect on skeletal muscle spasm in the lumbar region and neck: two double-blind controlled clinical and laboratory studies. Comparative efficacy and safety of skeletal muscle relaxants for spasticity and musculoskeletal conditions: a systematic review. Comparative outcomes of a 2-year follow-up of cervical medial branch blocks in management of chronic neck pain: a randomized, double-blind controlled trial. Cervical medial branch blocks for chronic cervical facet joint pain: a randomized, double-blind, controlled trial with one-year follow-up. Effectiveness of cervical medial branch blocks in chronic neck pain: a prospective outcome study. Long-term follow-up of patients treated with cervical radiofrequency neurotomy for chronic neck pain. M is a 54-year-old man referred to the pain clinic by his general practitioner with the request to perform an epidural corticosteroid infiltration at cervical level. The patient has a history of 2 months of right-sided neck pain radiating over the shoulder into the lateral forearm and index finger. Plain X-rays of the neck indicate degenerative facet (zygapophyseal) joints, predominantly at C3. The patient received instructions for exercises (rolling neck movements and arm rotation). The patient claims having done these exercises, but pain during the exercise discourages him to continue. The efficacy of this intervention has been demonstrated in subacute cervical radicular pain. The pain specialist must confirm the diagnosis prior to planning any intervention. During history taking, indications suggestive for vascular diseases, tumor, or infection, the so-called red flags, should imperatively lead to further investigations that exclude serious pathologies. This type of patient may experience improvement with cognitive behavioral therapy. When red and yellow flags are excluded, attention is directed toward the pain pattern. Patients suffering cervical radicular pain report pain in the neck that radiates over the posterior shoulder into the arm and sometimes into the hand. Radicular pain is a symptom that is caused by ectopic impulse formation, whereas radiculopathy also includes neurologic signs such as sensory or motor changes. Epidural corticosteroid infiltration has the objective of administering the corticosteroid close to the inflamed nerve 258 the dermatomal and dynatomal maps are distinctly different. Pain originating from C5 radiates up to the upper arm, and pain from C6 and C7 radiates from the neck to the shoulder, forearm, and hand. The pain covers the posterolateral side of the upper arm, but the pain from C7 extends more dorsally. For the Spurling test, the neck is extended with the head rotated to the affected shoulder while axially loaded.
Tissue biopsy from affected sites will show a greater proportion of cells with the mutation while biopsy from normal appearing sites will have mostly genetically normal cells bacteria nucleus purchase 100 mg azrolid with mastercard. Oligogenic Inheritance While monogenic disorders have been classically described to be caused by mutations within a single gene, of late some so called Mendelian disorders have been found to be caused by simultaneous mutations within two or more loci, a phenomenon referred to as oligogenic inheritance (oligogenic meaning caused by a few genes). Multifactorial Inheritance Disorders which are caused by a complex interaction of many different genes and environmental influences are called multifactorial disorders. Individual alleles involved in the causation of multifactorial disorders may individually follow any of the Mendelian or non-Mendelian inheritance patterns described above, but the disorder per se, which is a composite manifestation of the different alleles, follows a complex inheritance called multifactorial inheritance. Multifactorial disorders include congenital malformations, such as neural tube defects, cleft lip, cleft palate and congenital hypertrophic pyloric stenosis, as well as adult onset disorders such as diabetes mellitus and coronary artery disease. Phenomena like oligogenic inheritance, reduced penetrance and variable expressivity suggest that even the so called monogenic disorders are significantly influenced by other genes and environmental factors. As our understanding of molecular mechanisms of genetic diseases improves, conventional concepts about inheritance patterns are likely to significantly change in the future. Non-Mendelian mechanisms of inheritance include trinucleotide repeat expansion, mitochondrial inheritance, genomic imprinting, uniparental disomy, mosaicism, oligogenic inheritance and multifactorial inheritance. Once the pattern of inheritance is known, one can predict the risk of recurrence of the disorder in offspring, siblings and other relatives of the index case and offer appropriate interventions such as prenatal diagnosis and presymptomatic diagnosis. However, while the classical textbook descriptions of Mendelian and non-Mendelian inheritance patterns are still relevant and useful especially in the clinical setting, the strict distinction between monogenic and Chapter 2. This article deals with the approach to diagnosis of syndromes associated with developmental abnormalities of body structure. David Smith defined dysmorphology in 1966 as a study of abnormality of body structure that originates before birth. Reardon and Donnai in their review note that no branch of science offers as much opportunity, challenge and excitement as is there in the diagnosis of a rare disorder or a syndrome in clinical genetics. However, it is not a profession of stamp collectors but one that affords affected families with an answer why their child is different, anticipates and treats complications, and prevents recurrence in affected families. A three generation family history and consanguinity history are important because if present, inheritance patterns can be ascertained (Case 1). Parental ages at conception, teratogen exposure, history of abortions and stillbirth or abnormalities in the fetus in previous conceptions should be noted. History of maternal disorders like diabetes and infections during pregnancy should be taken Table 1). Prenatal ultrasonography records may provide information about abnormal growth pattern, oligoamnios or polyhydramnios. Events at birth, including fetal distress, birthweight, length and head circumference, and neonatal behavior and feeding history should be sought. Details of development history and behavior with formal assessment are very important. A look into the old records, noted findings and treatment as well as anthropometry details with growth charting over time can assist to reach a conclusive diagnosis in some cases. The first child, female had mild intellectual disability and she was not dysmorphic on examination. On further questioning, the mother of the children was slow in her daily tasks and thinking abilities, though she had completed her graduation. Her father too was reported to be slow intellectually, but was employed and never required assistance for daily living. On direct questioning his maternal uncle was not as bright as his peers and other sibling and had no children. The females are relatively mildly affected and the phenomenon of anticipation is apparent. The behavior phenotype, subtle dysmorphism and the inheritance suggests a diagnosis of the commonest X-linked mental retardation syndrome, fragile X mental retardation syndrome. There are definite implications for management, anticipation, evaluation and treatment of complications like atlantoaxial dislocation in Larsen syndrome as well as prevention of recurrence in subsequent pregnancies. It also provides answer to the family why their child is different and halts a diagnostic odyssey and frustration of endless consultations with doctors.
The other set of disorders that occur due to mutations on X-linked genes are the X-linked dominant disorders 2013 generic azrolid 500mg on-line. Like dominant mutations on autosomal genes, dominant mutations on X-linked genes are also expressed in heterozygotes and therefore heterozygous females as well as hemizygous males are affected. However, due to the phenomenon of lyonization (random inactivation of one of the two X chromosomes in each cell in females), females are usually less severely affected than males. An affected father will transmit the disorder to all his daughters but to none of his sons as male to maletransmissionofX-linkedmutationsdoesnotoccur. FewmoreexamplesofX-linkeddominantdisorders are X-linked hypophosphatemic rickets (vitamin D resistant rickets), incontinentia pigmenti, orofaciodigital syndrome type 1, Goltz syndrome (focal dermal hypoplasia) and Melnick-Needles syndrome. Several X-linked disorders, such as fragile X syndrome and ornithine transcarbamylase deficiency (a type of urea cycle defect), cannot be strictly categorized as X-linked recessive or dominant conditions, because they have very variable expression in female individuals. These can lead to manifestations of X-linked recessive disorders and suppression of the phenotype of X-linked dominant conditions in females. Till date, no single-gene disorder has been identified to have Y-linked inheritance. Hairy ears and a type of deafness (Y-linked deafness) were believed to be associated with genes on the Y chromosome, but studies disproved this. These microdeletions can be transmitted from an affected male to his male offspring, if his sperms are used in assisted reproductive techniques such as intracytoplasmic sperm injection. There can be further expansion in the number of repeats during gametogenesis resulting in the affected offspring having a larger repeat number compared to the affected transmitting parent. The increasing number of repeats leads to increasing severity and earlier onset of the disease in successive generations-a phenomenon known as anticipation, which is the hallmark of trinucleotide repeat disorders. For some disorders, the number of repeats can increase so much during gametogenesis that the offspring can get a very severe form of the disease. Mitochondrial Inheritance Disorders caused by mutations in genes in the mitochondrial genome show a mitochondrial inheritance pattern. Each cell has a few to several thousand mitochondria within it and each mitochondrion contains dozens of copies of the mitochondrial genome. Mitochondrial mutations may be inherited or they may be acquired and accumulate during mitotic cell divisions. A given mutation may be present in only some of the mitochondrial genomes within a mitochondrion and in only a proportion of the mitochondria within a cell, leading to a heterogeneous population within an individual, within a tissue, within the same cell and even within the same mitochondrion, a condition referred to as heteroplasmy. If the mutation is present in all or at least a majority of the mitochondrial genomes of an individual, the condition is called homoplasmy. Expression of a mitochondrial disorder in an individual and in each body tissue occurs only when the number of mutated mitochondrial genomes in that tissue exceeds a baseline, a phenomenon referred to as threshold expression. As only the egg cell contributes mitochondria to the zygote, the mitochondrial mode of inheritance is strictly maternal and affected males do not pass on the mutation to their offspring. However, the proportion and distribution of abnormal mitochondrial genomes can vary remarkably, resulting in great variability in the clinical severity amongst the affected individuals in a family. Some disorders caused by mutations in the mitochondrial genome include Leber hereditary optic neuropathy, chronic progressive external ophthalmoplegia, some types of Leigh disease, mitochondrial encephalomyopathy with lactic acidosis and strokelike episodes syndrome, myoclonic epilepsy with ragged red fibers syndrome, Kearns-Sayre syndrome and neuropathy, ataxia and retinitis pigmentosa syndrome. Mitochondrial diseases usually affect organs with greater energy utilization and with greater rates of oxidative phosphorylation. Imprinting Related Disorders Genomic imprinting refers to the differential expression of alleles of certain genes based upon their parental origin. For such genes, only one of the two alleles, either the paternally derived one or the maternally derived one, would be actively expressed, while the other allele would be silenced through epigenetic mechanisms such as methylation. Therefore, a mutation will have phenotypic effects only if it is present on the active allele but not if it is on the silenced allele. For example, if for a given gene the maternally derived allele is imprinted, then a female affected with a diseasecausing mutation in that gene will have phenotypic manifestations of the disorder but her offspring who inherit the mutation from her will not be affected. Uniparental Disomy Normally, of the two homologues of each chromosome in an individual, one is derived from the father and one from the mother. Occasionally, both the chromosomes may be derived from the same parent, a condition referred to as uniparental disomy. Azrolid 500 mg low cost. HUGE Amazon Leggings & Activewear TRY ON HAUL | Cheap AMAZON Workout Clothes | Danielle McElroy. Use of cornstarch feeds help in maintaining blood glucose for longer periods of time and decreasing episodes of hypoglycemia antibiotics no dairy discount azrolid 250 mg on-line. The regions of brain commonly affected by hypoglycemia are parieto-occipital region, hippocampus, caudate nucleus and putamen. Prolonged, recurrent and severe symptomatic hypoglycemia is associated with long-term neurologic sequelae and intellectual disability, visual deficits, motor deficits manifesting as spasticity or ataxia, seizure disorder and even microcephaly. In a study done at a tertiary care center in south India, neonatal hypoglycemia was found to be the most common etiology of remote symptomatic infantile onset epilepsy. Babies with symptomatic hypoglycemia have a poorer prognosis than those with asymptomatic hypoglycemia. In babies with hypoglycemia related to inborn errors of metabolism, survival depends on the severity of the underlying defect. Ketotic Hypoglycemia the treatment of ketotic hypoglycemia consists of frequent feedings with a high-protein and carbohydrate diet. During intercurrent illnesses, urinary ketones (which usually precede hypoglycemia) should be monitored. In the presence of ketonuria, if the child is accepting orally, liquids of high carbohydrate content should be offered to the child. In children with fatty acid oxidation defects, carnitine supplementation has been found to be beneficial. In phosphoenol pyruvate carboxykinase deficiency, avoidance of periods of fasting through frequent feedings rich in carbohydrate should be helpful, because glycogen synthesis and breakdown are intact. Hyperinsulinism is the most common cause of persistent hypoglycemia in early infancy, for which the most common underlying etiology is persistent hyperinsulinemic hypoglycemia of infancy, while in older children, ketotic hypoglycemia is most common. The treatment comprises of management of hypoglycemic episodes, changes in the dietary pattern and use of drugs based on the underlying etiology. Prolonged, recurrent and severe symptomatic hypoglycemia is associated with long-term neurologic sequelae. Neonatal hypoglycemic brain injury-a common cause of infantile-onset remote symptomatic epilepsy. Knowledge gaps and research needs for understanding and treating neonatal hypoglycemia: workshop report from Eunice Kennedy Shriver National Institute of Child Health and Human Development. Abdominal pain mimics many surgical and medical conditions and diagnosis is often delayed or never made. In addition to the clinical features similar to many common disorders, other symptomatologies, mode of inheritance, intermittent presentations and decreased penetrance make this group of genetic metabolic disorders stand out. Porphyrias are a group of eight inherited disorders of heme synthesis, each caused by deficiency of a specific enzyme in heme synthesis pathway. These pyrrole rings are interconnected through methane rings between their a-carbon atoms. Porphyrins containing ferrous ion in the center are called heme and heme containing proteins are called hemoproteins. Examples of hemoproteins are oxygen carrying molecules (hemoglobin, myoglobin), cytochromes P450 which are involved in the metabolism of many drugs, cytochromes b and c are involved in oxidative phosphorylation, catalase, peroxidase and endothelial nitric oxide synthase. Block in the synthesis of heme causes accumulation of its precursors as well as deficiency of necessary heme-containing products. The symptomatic porphyrias are more common in females and pan-ethnic in distribution. The combined prevalence is around 5 in 100,000 with highest prevalence in Sweden (1 in 10,000). Each of these four acute hepatic porphyrias is caused by deficiency of distinct enzyme in heme biosynthesis pathway Table 1), but clinical signs and symptoms are the same. Most people affected with the disorder develop symptoms in second or third decade and rarely after menopause. Because of reduced penetrance and nonspecific symptoms, high suspicion is to be kept in mind to make a diagnosis. In skin, these accumulated porphyrins absorb light and convert to their ground state by transferring their energy to other molecules like membrane lipids, nucleic acid and other proteins and is the cause of photosensitivity. The enzymes, their properties and a brief description of the type of porphyria associated are described in Table 1.
|